04 Steps to Investigate Out of Specification (OOS) Result
- Published on: Mar 08, 2024
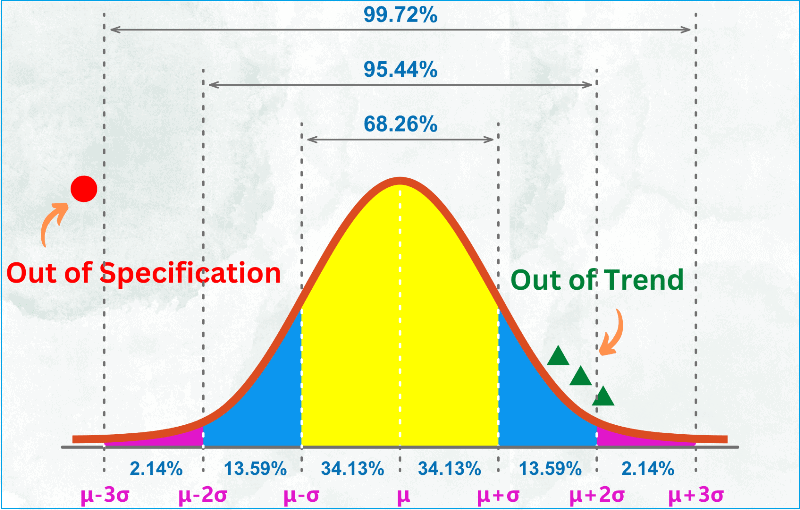
What is out of specification (OOS) result?
An Out of specification (OOS) result is simply the result that lies outside the specifications to which it is required to conform.
When you conduct a quality control test on a finished product or raw material and your test result falls outside of approved, registered, or official specifications, you should assign it an Out of Specification result and commence a rigorous investigation.
A confirmed out of specification (OOS) result leads to a non-conforming product.
What is the difference between OOS and OOT?
OOT stands for out-of-trend result.
When you conduct a quality control test on a finished product or raw material and your result falls within the official specifications but is atypical based on experience or empirical data, you may call it out of trend result.
Another way to describe a test result that does not follow the expected trend for a particular batch or series of batches is by comparing it with previous results.
Out of trend results are not yet to be called out of specification but require your attention to study the situation to prevent a potential out of specification result in the future.
It simply alerts you that something is fluctuating and may cause out of specification down the line.
Typically, OOT results would fall outside the alert limit but within the action limit.
There are three types of Out of trend situations:
– Atypical result: A single result that does not follow the expected trend compared to previous results from the same study
– Atypical trend: A set of test results showing an atypical pattern. Detecting an atypical trend requires knowledge of the expected trend, i.e., the typical trend of stability data from other studies on the product.
– Adverse trend: A series of test results showing a decrease/increase that indicates a probability of an out of specification result.
 Result 1")
What is the origin of out of specification events?
In the early 1990s, the Food and Drug Administration (FDA) inspected a major generic manufacturer in the US and identified concerns with failing test results.
In the FDA’s view, the company was not thorougly investigating Out of Specification (OOS) results but was merely re-testing the product for compliance.
The manufacturer challenged the FDA’s view, ending in the US District Court.
The court ruled that any individual OOS event should be investigated, and if a laboratory error cannot be identified, the batch will fail the test.
This case established the universal rules or principles for the treatment of OOS conditions:
– Conduct an initial informal laboratory investigation.
– If you need to, please conduct a formal management investigation.
– Please make sure that proper documentation is maintained and corrective action is undertaken.
The FDA subsequently published several guidance documents regarding this matter.
The current guidance provides clear requirements for investigating and resolving an out-of-specification incident. All regulators use the principles behind this guidance.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
How to determine out of specification results
Suppose you have tested 20 samples from a recently produced tablet batch. Each tablet has the label claim of 100mg (w/w).
It is improbable that all 20 results will be 100 mg exactly. Test results show that some of your data are above that target and below.
How do you determine if there is an Out of Specification and Out of Trend?
– You will plot all 20 test results as shown in the spreadsheet below.
– Calculate the Mean and Standard Deviation (σ). Using these two, calculate the control limits and specification limits.
– Control limits are inherent process variability, while specifications are set based on customer requirements.
– The upper control limit (UCL) is the maximum process variation value (tablet compression machine).
– You can calculate UCL as Mean + 3 x Standard deviation (σ).
– Similarly, the lower control limit (LCL) is the minimum value of process variation.
– Calculate LCL as Mean – 3 x Standard deviation (σ).
– Typically, specification limits are set narrower than control limits. However, this is based on your confidence in the process capability.
– In this case, you have set the specification limits as 100mg + 2 mg. Hence, the USL is 102 mg, and LSL is 98 mg.
– Change some values in column C on the spreadsheet below and find if you have Out of Specification results. The Range chart display will change based on your inputs.
– You can also determine if there are Out of Trend results. This occurs when five successive data are above or below the target value.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
What are the typical sources of out of specification events?
Out of specification results can be derived from anywhere in production or quality control facility where tests are carried out and matched against approved specifications.
The following are common sources or categories of OOS events:
Category 1: Laboratory error
Laboratory errors include analyst errors, incorrect calculations, malfunctioning equipment, incorrect standards or sample preparation, and measurement errors.
Confirmation that this caused the OOS condition would not constitute a product failure.
Laboratory errors should be relatively rare. Frequent errors suggest a problem that might be due to inadequate analyst training, poorly maintained or improperly calibrated equipment, or careless work.
Whenever a laboratory error is identified, the laboratory should determine its source and take corrective action to prevent recurrence.
Category 2: Lack of method precision
In the original legal ruling regarding OOS conditions, this category was not recognized since it was assumed that all validated test methods should have adequate precision.
Individual results may fall outside the specifications by chance alone due to inherent variation within the assay.
Confirmation that this was the cause of the OOS condition does not constitute a product failure.
However, it would indicate that the validity of the test method is questionable due to a lack of precision.
Category 3: Non process-related or operator error in manufacturing
This category is concerned with human or mechanical errors that occur during the manufacturing process.
For example, failure to add a component, operator error, malfunction of equipment, or cross-contamination. Confirmation that this was the cause of the OOS condition would constitute a failure of a specific batch of product.
Category 4: Process or manufacturing problem
This category concerns process or manufacturing errors due to control over processes.
For example, incorrect mixing times and heterogeneity of blends.
Confirmation that this caused the OOS condition would constitute a failure within that particular lot, but it may also mean that other lots may be impacted as potential failures.
How do we investigate out of specification (OOS) results?
Regulations are very sensitive about how test results that are out of specification should be conducted.
Pharmaceutical manufacturers must have written procedures on the steps to take when any result does not meet specifications.
Out of specification (OOS) rules require that any single result that does not meet specification must be investigated and not discarded without written justification or evidence that it was a genuine analyst error.
In addition, simply averaging a failed result and passing the result to obtain an average passing result could be interpreted as “testing into compliance.”
All OOS events must be looked into and resolved quickly. All investigations, conclusions, decisions, and corrective actions must be documented and retained as part of the official laboratory records for that particular lot.
 Result 2")
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
Step 1: Understanding assignable cause in out of specification investigation
Before conducting an out of specification investigation, you should understand assignable causes and whether an assignable cause has resulted in the out of specification event.
Test results, whether in or out of specification, obtained under the following assignable causes must be invalidated, and the test should be repeated.
– Sample: the original sample was not representative or was insufficient in quantity.
– Method/Documentation: the directions for the test method or Standard Operating Procedure (SOP) were unclear, resulting in incorrect test execution.
– Analyst Error: examples include, but are not restricted to, incorrect weight of sample used, sample or sample solution spills, dilution errors, or improper test procedure; and
– Instrument/Mechanical/System Malfunction: examples include, but are not restricted to, interfering electrical surges or spikes, the pump stops pumping, or the injector stops injecting.
If the Initial Investigation determines a readily apparent assignable cause, the initial out of specification result is invalidated.
Category 1 and 2 out of specification events are mostly to be attributed to these assignable causes.
Step2: Conduct initial out of specification investigation
The exact cause of an out of specification event can be difficult to determine with certainty, and it is unrealistic to expect that analyst errors will always be documented.
Nevertheless, an initial laboratory investigation must include more than a simple repeat or retest.
Simply retesting as a strategy raises three problems:
– It is a regulatory requirement to investigate before any retests are conducted.
– It may imply a lack of control. The laboratory is not concerned about the causes of possible failures, and therefore, these conditions may recur on retest.
– It may lead to the discarding of the original result without invalidation
Therefore, a laboratory must predetermine its course of action in an out of specification event.
This should be done using a combination of standard operating procedures supported by an OOS investigation form.
This approach minimizes errors and subjective decisions and ensures the investigation is thorough and fully documented.
The initial investigation includes, but is not limited to, examinations of the following:
– Analytical records (raw data/laboratory notebooks),
– Analytical procedures used,
– Formula and Calculations used,
– Equipment or Instruments used,
– Sample preparations, storage and handling,
– Testing environment, and
– Analyst error
You are right if some of the initial investigational areas above look like the assignable causes in step 1.
That’s because the objective of an initial investigation is to ascertain if an assignable cause is readily apparent that might be attributed to an out of specification result.
In that case (an assignable cause is found without a doubt), declare the original out-of-specification result invalid and repeat the test to replace the invalidated result.
Step 3: Conduct a formal out of specification investigation and measurements
If the out of specification result cannot be invalidated by the initial investigation, or if there are multiple OOS events, you should initiate a full-scale formal inquiry involving management, QA and QC personnel.
Multiple OOS results would indicate that the cause of the problem is most likely to be either Category 3 or 4 events.
Right now, you should extend the investigation beyond the laboratory to the production area.
The batch record and any batch processing deviations should be reviewed to formally identify the source of the deviation from specification.
If you do not have sufficient original samples on hand, you must decide to resample at this point of the investigation.
The formal investigation is conducted to identify process—or non-process-related errors.
Ultimately, QA management will review the investigation, draw conclusions, and propose corrective actions.
Some requirements for formal out of specification investigation
Formal investigations extending the laboratory (Category 3 or 4) must follow an outline or protocol with particular corrective action.
The investigation team must follow the steps bellow:
1. State the reason for the investigation
2. Summation of the process sequences that may have caused the problem
3. Outline corrective actions necessary to save the batch and prevent similar recurrence
4. List other batches and products possibly affected, the result of an investigation of these batches and products, and any action. Specifically:
– Examine other batches of product made by the errant employee or machine
– Examine products produced by the errant process or operation
5. Preserve the comments and signatures of all production and quality control personnel who conducted the investigation and any reprocessed material after additional testing.
The outcome of the formal out of specification investigation leads you to one of the following considerations:
– If an assignable cause is identified due to the formal investigation, you can invalidate the initial OOS result. The original testing is repeated to generate valid results; or
– If no assignable cause is identified as a result of formal investigation, the investigation team must evaluate the need for raising an Alert.
Where a sufficient sample is available, you should initiate a retest of the same sample under a retest protocol.
Step 4a: Repeating the test (when an assignable cause is identified)
When an assignable cause is identified by the results of either the initial or formal out of specification investigation and measurements, the original OOS result is invalidated.
In this case, you should repeat all the invalidated tests. The result will replace only the original invalidated results.
If the assignable cause was not due to the sample preparation, the original sample preparation(s) shall be used for this testing.
A single analysis replaces a single initial out of specification result.
For example, an assignable cause has been identified for one of the ten results from a Content Uniformity test or one of the six results from a dissolution test.
The original single result is invalidated.
To replace the single invalidated result, you should test one additional dosage unit for the content uniformity test or perform a dissolution test using one dosage unit.
Step 4b: Conduct a retest (when no assignable cause is identified)
You should conduct a retest under a carefully designed retest protocol based on the specific problem identified, the product history, the method, and the batch.
You must delineate the number of retests to be performed.
The retest actions resulting from an OOS investigation follow some important rules.
– A retest is defined as additional testing on the same sample (from the same bottle of tablets or capsules and the same drum or mixer). Retesting is acceptable only after the investigation has commenced and only if retesting is appropriate to investigate the OOS event.
– If you have enough samples available, the retest protocol must be executed using the same sample set that was the source of the original out-of-specification result.
– You should adopt the rule that a minimum of three (3) retests is required for all types of samples, except a minimum of five (5) retests is required for formulated products (i.e., In-Process and finished product samples).
– A Control sample may be used to verify the accuracy of the analyses. The retest protocol must specify acceptance criteria for the control sample.
– Consider assigning an analyst other than the one who performed the original test to perform retesting.
– You cannot continue retesting indefinitely. The retest protocol should nominate the number of retests required.
At the conclusion of retesting, a decision should be made to either accept or reject the batch.
Additional retesting should not be conducted to ‘test the product for compliance’.
FDA guidance on retesting as a result of out of specification investigation
– A retest is acceptable if reviewing the analyst’s work indicates an analyst’s error (Category 1).
In this case, limited retesting is required, and the retest result may replace the original result.
– A retest is acceptable if the investigation is inconclusive as to whether the OOS is category 1 or 2, as the laboratory needs to determine whether the OOS result is an outlier or a reason to reject the batch.
– Retest results should be used to supplement initial results in Category 2.
– Resampling is appropriate where provided for by an official monograph, such as sterility testing, content uniformity, and dissolution testing.
– Resampling is acceptable in the limited circumstances in which an OOS Investigation suggests that the original sample is unrepresentative.
Evidence, not mere suspicion, must support a resample designed to rule out preparation error in the first sample.
(Ref: FDA inspection Guide, Pharmaceutical Quality Control Labs (7/93))
 Result 3")
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
How do we evaluate retest results in out of specification investigation?
As you know from the earlier section, if an assignable cause is found to be associated with the out-of-specification result, the original result is invalidated.
If no assignable cause is found and retesting is performed, all test results shall be documented on the retest protocol.
All retest results must conform to specifications to overcome the initial OOS result for raw materials, intermediates, and API Samples.
– If all results obtained are within specification, then the mean of the retest results shall be reported as the final valid result or
– If any of the results obtained during retesting are outside the specifications, then the original OOS result is confirmed and reported as the final valid result.
For formulated products (i.e., in-process and finished product samples) and medical devices, assess the retest result by following rules:
– If all retest results are within specification and the original OOS result is outside three standard deviations of the mean of the retest results, then the average of the retest results shall be reported as the final valid result; OR
– If any results obtained during retesting are outside specifications or the original OOS result is within three (3) standard deviations of the mean of the retest results, then the original OOS result is confirmed and reported as the final valid result.
Averaging of final retest results during the investigation
In general, relying on the average figure without examining and explaining the individual OOS results is highly misleading and unacceptable to regulatory bodies.
Although averaging test data can be used to summarise results, laboratories should avoid reporting only averages because averages hide the variability among individual test results.
Final results should be reported including all individual results and the average result. Management should review individual results before releasing batches for sale.
Averaging is particularly troubling if testing generates both OOS and passing individual results, which, when averaged, are within specification.
This may occur in the case of Category 2 conditions.
There are both appropriate and inappropriate uses of averaging test data during original testing and an OOS investigation.
(Note: In certain circumstances, principally for biological assays, Individual monographs may allow the reporting of average results alone.)
What documentation is required during an out of specification investigation?
One of the most important aspects of any investigation, whether informal in the laboratory or formal by management, is the requirement to fully document each step of the investigation and any conclusion found.
Regulatory authorities may review the documentation at a later stage to determine whether the company is deliberately discarding or ignoring aberrant results.
The documentation trail must start once the out-of-spec result is detected and before any action is taken.
The best way to control this is to establish clear rules and regulations for analysts and follow up with support through training, an audit, and a standard investigation report form.
Corrective action steps and management decisions should also appear on the investigation record, along with an assessment of whether any other tests have been compromised.
In summary, the OOS documentation needed for the investigation should include:
– A standard operating procedure that predetermines the laboratory’s course of action
– A standard form for analyst investigation
– An authorized investigation report
– An out of specification trend record/register
Analysts’ mistakes, such as undetected calculation errors, should be specified with particularity and supported by evidence.
Investigations and conclusions must be preserved with written documentation that enumerates each step of the investigation.
The evaluation, decision, and corrective action, if any, should be maintained in an Investigation or failure report and placed into a central file.
(Ref: FDA inspection Guide, Pharmaceutical Quality Control Labs (7/93))
Conclusion
We may occasionally encounter results that are out of specification for various reasons.
An out of specification event must be addressed as quickly as possible through initial investigation to determine whether any assignable cause, such as analyst error, instrumental error, test method precision, or calculation error caused it.
If an assignable cause is found responsible for the out-of-spec event, the original OOS should be invalidated or ruled out.
If an assignable cause could not be attributed, a formal investigation must be undertaken beyond the laboratory environment.
Repeat testing or retesting of the same sample should determine if the original OOS result is confirmed or not.
It is important to keep all documentation generated during out of specification investigation for future regulatory audits. A written investigation record should include the following Information.
– A clear statement of the reason for the Investigation.
– A summary of the aspects of the manufacturing process that may have caused the problem.
– The documentation review results, with the actual or probable cause assignment.
– The review results were made to determine if the problem had occurred previously.
– A description of corrective actions taken.
No data or test result shall be excluded from final calculations unless it can be shown that there is a valid reason for exclusion.
Any exclusion must be documented with full justification and with the written approval of the QA Manager or Laboratory Manager.
240 SOPs, 197 GMP Manuals, 64 Templates, 30 Training modules, 167 Forms. Additional documents included each month. All written and updated by GMP experts. Checkout sample previews. Access to exclusive content for an affordable fee.
 Result 4")
Author: Kazi Hasan
Kazi is a seasoned pharmaceutical industry professional with over 20 years of experience specializing in production operations, quality management, and process validation.
Kazi has worked with several global pharmaceutical companies to streamline production processes, ensure product quality, and validate operations complying with international regulatory standards and best practices.
Kazi holds several pharmaceutical industry certifications including post-graduate degrees in Engineering Management and Business Administration.
Related Posts

How to Develop Quality Control Method and Specifications for a Laboratory
Disposition of Production Materials and Finished Products at a GMP Site
Basic cleaning and sanitation practices in Pharmaceuticals









Hello i think that i saw you visited my weblog so i came to Return the favore Im trying to find things to improve my web siteI suppose its ok to use some of your ideas
Good morning, I’m doing related to your post and I have few questions to you. Where did you find informations for your blog post? In papers, maybe in rags or books, or just randomly on the Web? Please respond :).
I sincerely took pleasure in reading your weblog, you explained some superior points. I want to bookmark your post. I saved you to delicious and yahoo bookmarks. I will attempt to revisit to your webpage and examine more posts.